
POMPE DISEASE:
Why in the News?
- Nidhi Shirol, India’s first Pompe disease patient, passed away at 24 after a prolonged battle with the condition, spending the last six years semi-comatose.
- Her father, Prasanna Shirol, established the Organisation for Rare Diseases India, the country’s pioneer NGO for rare diseases.
Source: Org. of Rare Disease India
 Source: Org. of Rare Disease India
Source: Org. of Rare Disease IndiaAbout Pompe Disease:
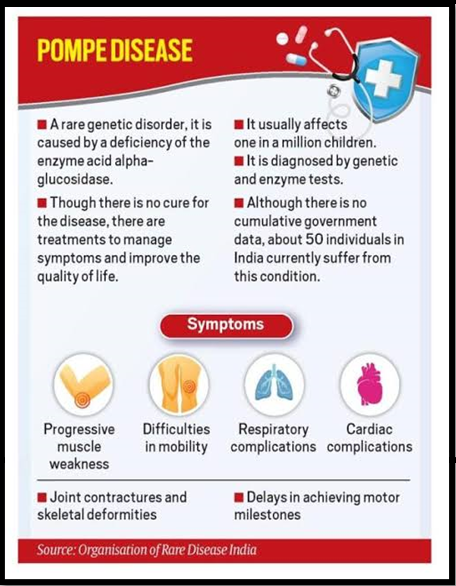
- Known as Glycogen Storage Disease Type II, Pompe disease is a rare genetic disorder caused by a deficiency of the enzyme acid alpha glucosidase (GAA), vital for glycogen breakdown in cell lysosomes.
- Prevalence estimates range from 1 in 40,000 to 1 in 300,000 births, showcasing varied onset ages and severity levels leading to diverse clinical manifestations.
Key Symptoms:
- Muscle Weakness: Progressive muscle weakness is prominent, affecting respiratory muscles and causing breathing difficulties, especially while lying down.
- Motor Skill Delays: Children might experience delays in achieving motor milestones like sitting, crawling, and walking.
- Respiratory Complications: Weakening respiratory muscles leads to breathlessness and infections.
- Hypertrophic Cardiomyopathy: Thickening of heart muscles can impair heart functions.
Diagnosis and Treatment:
- Diagnostic Methods: Enzyme assays measure GAA activity, while genetic testing identifies responsible GAA gene mutations crucial for confirmation.
- Management: Although no cure exists, treatments like Enzyme Replacement Therapy can enhance the patient’s quality of life by reducing glycogen build-up through enzyme infusion.
This succinctly outlines Pompe disease, its symptoms, diagnostic methods, and available management options, underscoring the importance of early detection and intervention despite the absence of a definitive cure.


